Joint Bayesian Estimation of Cell Dependence and Gene Associations in Spatially Resolved Transcriptomic Data
Abstract
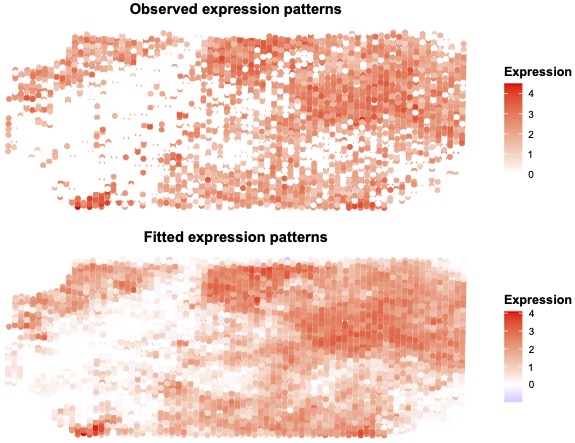
Recent technologies such as spatial transcriptomics, enable the measurement of gene expressions at the single-cell level along with the spatial locations of these cells in the tissue. Spatial clustering of the cells provides valuable insights into the understanding of the functional organization of the tissue. However, most such clustering methods involve some dimension reduction that leads to a loss of the inherent dependency structure among genes at any spatial location in the tissue. This destroys valuable insights of gene co-expression patterns apart from possibly impacting spatial clustering performance. In spatial transcriptomics, the matrix-variate gene expression data, along with spatial coordinates of the single cells, provides information on both gene expression dependencies and cell spatial dependencies through its row and column covariances. In this work, we propose a joint Bayesian approach to simultaneously estimate these gene and spatial cell correlations. These estimates provide data summaries for downstream analyses. We illustrate our method with simulations and analysis of several real spatial transcriptomic datasets. Our work elucidates gene co-expression networks as well as clear spatial clustering patterns of the cells. Furthermore, our analysis reveals that downstream spatial-differential analysis may aid in the discovery of unknown cell types from known marker genes.