Phylogenetically Informed Bayesian Truncated Copula Graphical Models for Microbial Association Networks
Abstract
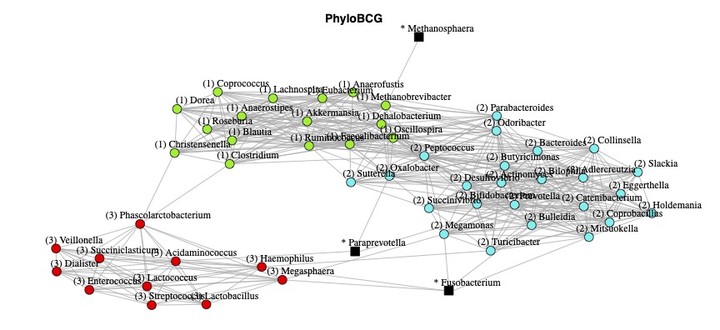
Microorganisms play critical roles in host health. The advancement of high-throughput sequencing technology provides opportunities for a deeper understanding of microbial interactions. However, due to the technological limitations of 16S ribosomal RNA sequencing, microbiome data are zero-inflated, and a quantitative comparison of microbial abundances cannot be made across subjects. By leveraging a recent microbiome profiling technique that quantifies 16S ribosomal RNA microbial counts, we propose a novel Bayesian graphical model that incorporates microorganisms’ evolutionary history through a phylogenetic tree prior and explicitly accounts for zero-inflation using the truncated Gaussian copula. Our simulation study reveals that the evolutionary information substantially improves the network estimation accuracy. We apply the proposed model to the quantitative gut microbiome data of 106 healthy subjects, and identify three distinct microbial communities that are not found by existing microbial network estimation models. We further find that these communities are discriminated based on microorganisms’ ability to utilize oxygen as an energy source.